Beta-Thalassemia Prevention and Control Program in Zahedan University of Medical Sciences, 2020
Beta Thalassemia is one of the most common inherited blood disorders and a major the public health problem across the world. Beta thalassemia is a blood disorder that reduces the production of hemoglobin. Hemoglobin is the iron-containing protein in red blood cells that carries oxygen to cells throughout the body. Beta thalassemia is characterized by the reduced synthesis (β+) or absence (βo) of the β-globin chains in the HbA molecule, resulting in accumulation of excess unbound α-globin chains that precipitate in erythroid precursors in the bone marrow and in the mature erythrocytes, leading to ineffective erythropoiesis and peripheral hemolysis.
Reportfrom: ZAUMS Non-Communicable Diseases Control Department
Published on: 28 August 2020
Beta Thalassemia is one of the most common inherited blood disorders and a major the public health problem across the world. Beta thalassemia is a blood disorder that reduces the production of hemoglobin. Hemoglobin is the iron-containing protein in red blood cells that carries oxygen to cells throughout the body. Beta thalassemia is characterized by the reduced synthesis (β+) or absence (βo) of the β-globin chains in the HbA molecule, resulting in accumulation of excess unbound α-globin chains that precipitate in erythroid precursors in the bone marrow and in the mature erythrocytes, leading to ineffective erythropoiesis and peripheral hemolysis.
Approximately 1.5% of the global population is heterozygotes (carriers) of the β-Thalassemia. The β-Thalassemia is more common among individuals originating from the Mediterranean area, Middle East, Transcaucasia, Central Asia, Indian subcontinent, and Southeast Asia. β-Thalassemia is classified into two types depending on the severity of symptoms: β-Thalassemia minor and β-Thalassemia major. Of the two types, thalassemia major is more severe.
Among the eastern Mediterranean region countries, Iran is one of the major centers for the prevalence of β-Thalassemia. Regarding to high consanguinity among population, it is estimated that there are between two and three million β-Thalassemia carriers and 25,000 patients in Iran. Like many other countries in the region, a large number of Thalassemia patients are β-Thalassemia.
Distribution of β-Thalassemia mutations in Iran shows different patterns in different areas. β-Thalassemia mutations have been a reflection of people and area in correlation with migration and origin of ancestors. The gene frequency of β-Thalassemia, however, is high and varies considerably from area to area, having its highest rate of more than 10% around the Caspian Sea, and Persian Gulf. The prevalence of β-Thalassemia gene in other areas is between 4% and 8%. The prevalence of β-Thalassemia gene in Sistan and Balouchestan has been estimated at 8.5%.
Since carrier frequency for β-thalassemia is alarmingly high in some parts of the country, a nationwide prevention program has been implemented that includes mandatory pre-marriage counseling with the aim of limiting carrier marriage. Interruption of the pregnancy is permitted for severe fetal disorders, including Beta Thalassemia, but only within the fourth month of gestation. Iran has made great progress in β-Thalassemia prevention and treatment. The β-Thalassemia prevention program was implemented in 1995 and the country-wide thalassemia treatment network, consisting of 64 medical universities and faculties, is active. The main prevention programs strategies comprise carrier detections, molecular diagnostics, genetic counseling, and prenatal diagnosis. Since 1999, the new technological methods for the prenatal diagnosis (PND) of the affected embryos were used for all people, both in rural and urban areas.
As a result of quality services and good management of cases, the average age of patients with beta thalassemia in Iran has increased from 15 years in 1997 to about 35 years in 2018. Similarly, due to provision of quality services and good management of cases, the average age of patients with beta thalassemia in Iran has increased from 15 years in 1997 to about 35 years in 2018.
Beta-Thalassemia in Sistan and Baluchistan Province and ZAUMS
According to the Sistan and Balouchistan Province Blood Transfusion Organization, there are currently 3200 Beta-Thalassemia patients in the province who have been receiving blood transfusion services from the blood transfusion centers of the province. Accordingly, approximately 60% of the blood in Sistan and Baluchistan is currently used for Thalassemia patients. The number of major thalassemia cases in the population covered by Zahedan University of Medical Sciences was 1142 cases by August 2020.
From 2014 to the end of the June 2020, a total of 233 affected pregnancies with Beta-Thalassemia major had been identified and referred in a timely manner, of which 210 cases (90%) have resulted in abortion.
Figure 1- The trend of incident cases of Beta-Thalassemia in ZAUMS, 2012-2020
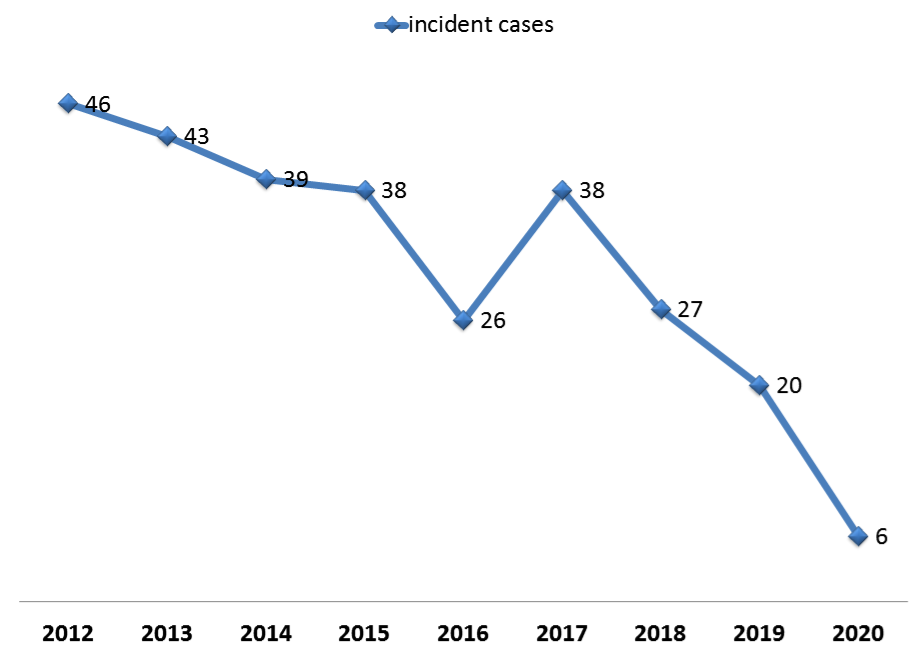
Among the most important measures taken in ZAUMS in the Beta-Thalassemia Prevention and Control Program, was a campaign for identification of carrier couples who had no history of Beta-Thalassemia screening tests in the rural and suburban areas, which was launched with the participation of Community Health Volunteers (CHVs) in 2016.
Free of cost services such as the possibility of prenatal diagnosis (PND) and medical termination of pregnancy if required, and free accommodation for poor couples in Zahedan, for performing diagnostic tests, performing a campaign to identify couples carrying Beta-Thalassemia, and holding Incident Cases Committees in cities covered by ZAUMS are part of other measures taken in the province to tackle the disease.
Premarital screening in adults is the ideal screening program as individuals can exercise choice in the selection of partners, understand the need for definite prenatal diagnosis, time may be gained by individuals to adjust to their carrier status, and pregnancy may be reported early. All those patients who turn out to be thalassemia carriers will be counseled and their partners screened for thalassemia. It is however not feasible in some parts of the province because of the custom of traditional marriages carried out before official registration. Although a religious consensus has been achieved on the abortion of fetuses affected by β-thalassemia from both Shia and Sunni clerics for issuing Fatwa permitting therapeutic abortion of affected fetuses, religious beliefs are still an important reason for opposing abortion. Some couples who opt to retain an affected fetus they are doing so based on religious restrictions and moral grounds, such as fear over committing a sin.
Providing families with information about the long-term effects of β-thalassemia on the child and family members can help them make informed decisions on pregnancy continuation or termination.
Copyright © 2020 Zahedan University of Medical Sciences. All rights reserved. Date Updated: 28/08/2020.
Should you have any queries please do not hesitate to contact us on: msartipi23@gmail.com











comment